


Advanced Heart Failure Team Offers Lifesaving Treatments
University Hospitals Harrington Heart & Vascular Institute provides lifesaving treatments for patients suffering from any stage of heart failure, including those who are critically ill, frail, elderly, and those who may be classified by other hospitals as “no option” patients.

Make a Referral
Call us at 216-844-3800 for more information or to refer a patient.Refer a Patient Today
Distinguished Site for Heart Transplant
Located at the University Hospitals Cleveland Medical Center, UH Harrington Heart & Vascular Institute is nationally recognized in the top 1% of nearly 5,000 hospitals as a Best Hospital for Cardiology & Heart Surgery by U.S. News & World Report. UH Harrington Heart & Vascular Institute is a distinguished site for heart transplantation, with certification by the Centers for Medicare & Medicaid Services (CMS). Our Ventricular Assist Device (VAD) Program is Join Commission Certified for meeting rigorous standards to support better patient outcomes.
UH Harrington Heart & Vascular Institute brings the latest medications, devices and stem cell therapies to its patients as a nationally selected site for various clinical trials.
UH Cleveland Medical Center Recognition

Joint Commission Certification of our Ventricular Assist Device (VAD) program for meeting rigorous standards to support better patient outcomes

Cardio-Oncology Program
Some forms of cancer therapies, including certain chemotherapy and radiation to the chest, can put cancer patients at an increased risk of having adverse cardiovascular events, particularly within the first five years. As one of a select few institutions globally, University Hospitals offers a Cardio-Oncology Program that provides comprehensive cardiovascular care for all cancer patients.
General Referral Guidelines
Learn more about when your patient should be considered for referral to our Advanced Heart Failure team.
Diagnosis & Treatment
Learn more about our specialized therapies to the unique needs of patients with chronic heart disease and heart failure.
Featured Articles & News
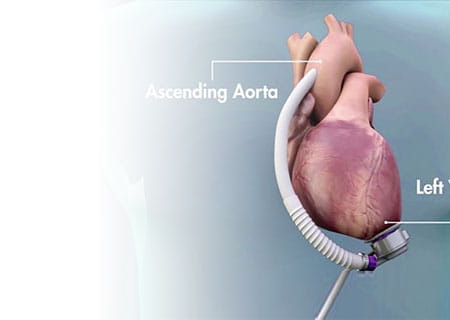
UH Cardiogenic Shock Team, Paired with LVAD Advancements, Changes the Odds for Shock Survivors
Patient Care
Learn more about how our Institute provides care for patients diagnosed with heart failure.
Download the MyUH App
Formerly the UH Provider App
